 |

01 Feb 2008
Recent technological advances in conjunction with major developments in fluorescent markers have made fluorescence microscopy an extremely powerful tool. René Hessling and Thorsten Kues of Carl Zeiss MicroImaging look at the techniques that are allowing researchers to study the structures within live cells in ever greater detail.
Fluorescence has become the most important contrasting method in light microscopy in recent years, largely thanks to the growing family of fluorescence proteins used for in vivo labelling.
Fluorescence microscopy provides the spatial, spectral and temporal resolution for monitoring dynamic processes inside living cells, intact tissue and even live animals. This article offers an overview of the powerful techniques currently used in light microscopy.
Entering 3D-space
In conventional fluorescence microscopy, an image consists of light gathered from the focal plane as well as from structures above and below this plane that are not in focus. This is a significant problem when studying thick specimens, where light emitted by the out-of-focus regions generates a substantial background signal. This is because neither the excitation light nor the emitted fluorescence is restricted to the optical axis of the microscope, which lowers the axial resolution and reduces the image contrast. 3D reconstructions with high spatial resolution are not possible from these wide-field images.
The solution to this physically inherent problem is optical sectioning. Optical sections contain only the information from the focal plane of the microscope and provide images with higher contrast and axial resolution. Today, several optical sectioning techniques are available, each with their respective strengths and drawbacks, but all of which have revolutionized microscopy during the last decade.
3D-deconvolution
Today’s wide-field fluorescence microscopes can acquire 3D-image stacks of multiple fluorescence channels with very high time resolution thanks to fast charge-coupled-device cameras and high-speed LED sources. However, as mentioned above, conventional wide-field systems cannot record optical sections directly.
After image acquisition, software can generate optical sections from the wide-field image stack using 3D-deconvolution (3D-DCV). This process uses information from the image formation process to reassign the out-of-focus signals back to the plane from which they originated. In contrast to alternative methods described below, 3D-DCV does not physically discard any light to generate high-resolution images and is therefore the most sensitive method available. When dealing with small and weak fluorescent structures, 3D-DCV is superior to most other optical sectioning techniques. Its major drawback is that optical sections are only available after the acquisition of entire 3D stacks and elaborate image processing.
Probing the surface with TIRF
Total internal reflection fluorescence (TIRF) microscopy is used to investigate how molecules interact with surfaces. TIRF involves using an evanescent wave to create a very thin optical section along the interface between the coverslip and the aqueous medium. The intensity of the evanescent field drops exponentially with penetration depth and results in an optical section thickness of around 100 nm. The technique only excites fluorescent molecules within this limited region, so TIRF images do not contain any background fluorescence and have superior contrast and signal-to-noise ratios. Photobleaching and phototoxicity are also reduced significantly. As a matter of principle, this optical sectioning technique is restricted to the coverslip-water boundary and cannot be used to acquire 3D image stacks. Nevertheless, it can be combined with 3D imaging on classical wide-field microscopes or confocal systems.
Confocal laser scanning microscopy
In confocal microscopy, optical sectioning is achieved by introducing an aperture diaphragm (pinhole) in a conjugated focal plane of the objective. This pinhole physically prevents light originating from above or below the focal plane from reaching the detector. Generally this principle only works for a diffraction-limited spot and acquiring an image involves scanning the focal point across the sample in both the x and y directions. These so-called point-scanning confocal microscopes provide high-resolution and high-contrast optical sections without the need for subsequent image processing. 3D image stacks can be acquired by recording a series of optical sections at different focus (Z) positions.
Recently, a variation of this technique has been developed where an entire line is illuminated and detected. To acquire an optical section, it is only necessary to scan the excitation line along the Y axis. This increases the scan speed dramatically and frame rates can reach up to 120 images a second at 512 x 512 pixel resolution. High-speed optical sectioning with line-scanning confocal systems is especially beneficial when observing dynamic processes in live cells.
Structured illumination
Another method for creating optical sections for high-resolution multichannel imaging is structured illumination. Here, the field stop in the excitation beam path of a conventional fluorescence microscope is replaced by a grid structure, which is projected into the focal plane of the objective. Three raw images are acquired with the grid structure in different positions and are subsequently combined in real-time to one optical section.
The principle of structured illumination is implemented as a simple insert for the field-stop plane. This can be installed easily and then upgraded for use in inverted and upright microscopes.
Structured illumination is particularly suited for 2D and 3D multichannel imaging of fixed specimen and for slow dynamic processes within living specimen.
Exploring the depth
Generally biological tissues are strongly scattering materials such that the deeper the target structure, the greater the absorption and scattering of visible light. Imaging structures several hundred microns deep within tissue at high spatial resolution is nearly impossible for wide-field and confocal microscopes.
This can be overcome with multiphoton microscopy, which uses near infrared laser pulses to excite the fluorescence. Near infrared light penetrates deeper into tissue and is generally less phototoxic. Similar to confocal point scanners, a focused laser beam is scanned over the specimen in the object plane and fluorescence only occurs if at least two photons are absorbed simultaneously by a fluorochrome.
The probability of excitation depends exponentially on the intensity of the excitation light, which is at a maximum at the objective focus. As a result, multiphoton excitation generates an intrinsic optical section. As in TIRF microscopy, there is no out-of-focus fluorescence excitation, which reduces phototoxicity and bleaching substantially. Fully motorized infrared lasers can be incorporated into confocal laser-scanning microscopes, adding the bene-fits of multiphoton imaging to confocal systems while maintaining ease of use.
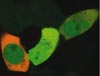
Experience fluorescence colours
Today, almost every protein can be tagged with fluorescent markers and visualized using fluorescence microscopy. One drawback is that the spectral properties of the available dyes can limit the experimental freedom. The goal is to distinguish clearly between the fluorescent markers in order to measure co-localization or functional interaction of the labelled proteins. However, this can be difficult even with two markers due to spectral crosstalk, and the problem grows increasingly complex with multiple labels.
One solution to this problem is a pro-cess known as “emission fingerprinting”, in which dyes are distinguished by their spectral charac-teristics. This method can be applied to wide-field fluorescence microscopes and confocal systems.
Certain confocal arrangements acquire a “lambda stack” for each image plane, essentially providing spectral information for each pixel of the image plane. Some pixels may contain the specific signal from just one of the fluorescent dyes in the sample, while others may contain a composite spectrum, which arises from two or more dyes that co-localize in the specimen.
Quantitatively separating these dyes is a three-step process. First, the raw data (lambda stack) is recorded. Second, the user specifies which dyes are to be expected in the sample by selecting reference spectra. Finally, a procedure named “linear unmixing” determines the relative contributions of the reference spectra to the recorded raw data in each pixel and creates images that display the relative contributions as intensities. The result is a clear and quantitative separation of fluorescent markers, even those that are spectrally very similar such as Sytox Green and FITC, or GFP and YFP. Examples have been published recently showing the clear separation of up to six fluorescent proteins expressed simultaneously in live cells.
Beyond imaging
Different fluorescence microscopy and optical sectioning techniques allow dynamic processes in live cells to be observed. In many situations proteins are found in an equilibrium state and are homogeneously distributed throughout the cell. In this instance, it is interesting to determine if these proteins are stationary because they are bound to the cytoskeleton or if they can diffuse freely throughout the cytoplasm. In order to measure these hidden dynamics, microscopy is used to optically manipulate the fluorescent distribution in the sample.
A classical experiment uses the focused laser beam in a confocal microscope to bleach the fluorescent signal in a defined region of the sample and then monitors the diffusion or transport of fluorescently labelled proteins back into this region. It is possible to measure the dynamic processes by monitoring the speed of fluorescence recovery after photobleaching (FRAP). Multiple variations of this technique such as inverse FRAP, fluorescence loss in photo-bleaching and fluorescence localization after photo-bleaching are routine applications in confocal microscopy today.
With the development of fluorescent proteins, such as PA-GFP, KAEDE or DRONPA, it is even possible to change the spectral characteristics of dyes by turning the fluorescent signals on or off or changing the colour of the marker by optically manipulating the sample.
• René Hessling is head of the training, application and support center and Thorsten Kues is an imaging and application specialist at Carl Zeiss MicroImaging GmbH. For more information see www.zeiss.de/micro.
• This article originally appeared in the January 2008 issue of Optics & Laser Europe magazine.
 |  |  |  |  |  |  |
| © 2026 SPIE Europe |
|





